HOMOZYGOUS DELETION IN THE SMN1 GENE IN ASYMPTOMATIC INDIVIDUAL - GENETIC COUNSELLING ISSUES IN SMA-RISK FAMILIES
BEZOBJAWOWE NOSICIELSTWO OBUALLELICZNEJ DELECJI W GENIE SMN1 A PORADNICTWO GENETYCZNE W RODZINACH RYZYKA SMA
Maria Jędrzejowska1, Krzysztof Szczałuba2, Danuta Sielska2
1Zespół Nerwowo-Mięśniowy, Instytut Medycyny Doświadczalnej i Klinicznej im. M. Mossakowskiego,
Polska Akademia Nauk
Kierownik: prof. dr hab. med. I. Hausmanowa-Petrusewicz
2Zakład Genetyki Medycznej, Instytut Matki i Dziecka, Warszawa
Kierownik: prof. dr hab. med. T. Mazurczak
AbstractSpinal muscular atrophy (SMA) is one of the most common autosomal recessive disorders. The mode of inheritance of SMA is what determines the relatively low risk to offspring of affected persons, sibs of carriers of the pathogenic mutation or their more distant relatives. Nonetheless, the risk in question is increased beyond that in the population in general, thereby indicating the need for preventative measures to be taken in respect of SMA families. We present clinical characteristics of such a SMA-risk family. An apparently unaffected brother (patient) of the proband together with his pregnant wife sought genetic counselling about the SMA risk in their offspring. The estimated a priori risk was of about 0.5% (1 in 200). Molecular diagnostic tests performed in the patient indicated the presence of a homozygous deletion of the SMN1 gene identical to the one detected in the affected proband. The patient’s wife was identified as a carrier of the deletion. The conditional risk for offspring of the couple was thus recalculated as 50% (1 in 2). Homozygous deletion of the SMN1 gene in unaffected individuals is a relatively rare event, yet one that nevertheless has significant impact on genetic counselling in SMA families. In these circumstances, molecular confirmation of SMA in such families allows for the provision of competent and reliable genetic advice, as well as for the introduction of secondary preventative measures. It also meets inclusion criteria as regards optional invasive prenatal diagnostic testing in families with a high (?25%) risk of the condition being present in the offspring.
Key words: SMA, asymptomatic homozygous SMN1 deletion, carrier, SMN2 copy number
Streszczenie
Rdzeniowy zanik mięśni (SMA) jest jedną z najczęstszych chorób o dziedziczeniu autosomalnym recesywnym.Ze względu na sposób uwarunkowania, ryzyko wystąpienia choroby u potomstwa osób chorych, rodzeństwa nosicieli patogennej mutacji oraz ich dalszych krewnych mieści się w granicach ryzyka małego. Jest ono jednak wyższe od populacyjnego i wskazuje na potrzebę objęcia rodzin SMA programem profilaktycznym. W pracy dokonano charakterystyki klinicznej rodziny ryzyka SMA. Klinicznie zdrowy brat chorego z SMA (pacjent) zgłosił się do Poradni Genetycznej wraz ze swoją ciężarną żoną w celu określenia ryzyka wystąpienia choroby u ich potomstwa. Ryzyko a priori oszacowano na około 0,5% (1/200). Przeprowadzone badania molekularne wykazały u pacjenta obecność obuallelicznej delecji genu SMN1, identycznej ze stwierdzaną u chorego brata. U żony pacjenta zidentyfikowano delecję w jednym allelu genu SMN1. Jest ona zatem nosicielką patogennej mutacji. Skorygowane ryzyko wystąpienia rdzeniowego zaniku mięśni u potomstwa pary wyniosło zatem 50%. Występowanie bezobjawowego nosicielstwa obuallelicznej delecji w genie SMN1 jest zjawiskiem rzadkim, istotnie jednak wpływającym na poradnictwo genetyczne w rodzinach ryzyka SMA. Przeprowadzenie badań molekularnych w rodzinach SMA umożliwia udzielenie pełnej i wiarygodnej porady genetycznej, z możliwością profilaktyki wtórnej. Stanowi również kryterium kwalifikacji do ewentualnej inwazyjnej diagnostyki prenatalnej w rodzinach obciążonych wysokim (?25%) ryzykiem wystąpienia SMA u potomstwa.
Słowa kluczowe: SMA, bezobjawowe delecje SMN1, nosicielstwo, liczba kopii SMN2
INTRODUCTION
Proximal childhood and juvenile spinal muscular atrophy belong to the most frequent genetic conditions. Among the autosomal recessive disorders, it constitutes the second cause of death of affected children, aft er cystic fibrosis. Spinal muscular atrophy is characterized by a progressive loss of spinal cord motoneurons that results in muscular weakness and atrophy. SMA frequency is estimated to be 1 in 7000 to 1 in 10 000 live births, refl ecting a mutated allele carrier frequency of 1 in 42 to 1 in 50 persons in the general population (1-5). Recent large case vs. control trials indicate an even higher carrier frequency of up to 1 in 25-37 individuals of Caucasian origin (6-10). Spinal muscular atrophy is characterized by significant phenotypic variability extending from congenital forms to asymptomatic cases. According to the International SMA Consortium guidelines, three main clinical forms of SMA are to be distinguished: the severe Werdnig-Hoffmann type (SMA1, children never sit up unsupported), the intermediate type (SMA2, children are able to sit unsupported but never walk) and the mild type (SMA3, muscular weakness appears aft er the ability to walk unaided is obtained) (11). Further reported clinical forms of spinal muscular atrophy are: congenital (symptoms in the fetus or shortly aft er birth) and adult (onset over the age of 20-30) (12, 13). There have also been reports of asymptomatic cases with typical SMA mutation (14, 15). All the existing forms of the disease are caused by mutations in the SMN1 (survival of motor neuron gene1) gene (16). Of these, over 95% (96.5% in the Polish population) refl ect biallelic (homozygous) loss (gene deletion or conversion) of exon 7 of the SMN1 gene (16, 17). In the remaining 2-3% of affected children, point mutations are identified on one allele, with the other carrying the usual deletion (18).
The main modifier of phenotypic variability in SMA is the SMN2 gene (19). It resembles SMN1 and encodes the same SMN protein. However, due to a single nucleotide change in exon 7, only about 10-20% of the SMN2 transcript translates into full-length SMN protein, while the other 80-90% devoid of the exon 7-encoded oligomerization domain is degraded. The SMN2 gene can exist in several copies. Healthy individuals are most likely to have one or two copies of the SMN2 gene (about 85% of the general population) (7, 20). In patients affected with SMA, this number can range from one to six. Children with the severe form usually carry one to three copies, with intermediate form three to four copies, and those with the mild or adult forms three to six copies (7, 21, 22). SMN2 gene copies are not functionally equivalent, since three copies have been observed in the severe as well as in the mild forms of the condition. This can most likely be explained by the infl uence of epigenetic modifiers, such as a differential pattern of DNA methylation (23)
In case of molecular confirmation of the clinical diagnosis of spinal muscular atrophy, a high-risk SMA family should be given appropriate genetic advice. In this paper, we present a family originally classified as low-risk of SMA. Molecular analyses in this family revealed the presence of a homozygous SMN1 gene deletion in an apparently asymptomatic patient, as well as a heterozygous deletion in his wife resulting in a 50% (1 in 2) risk of SMA occurring in the offspring.
PATIENTS AND METHODS
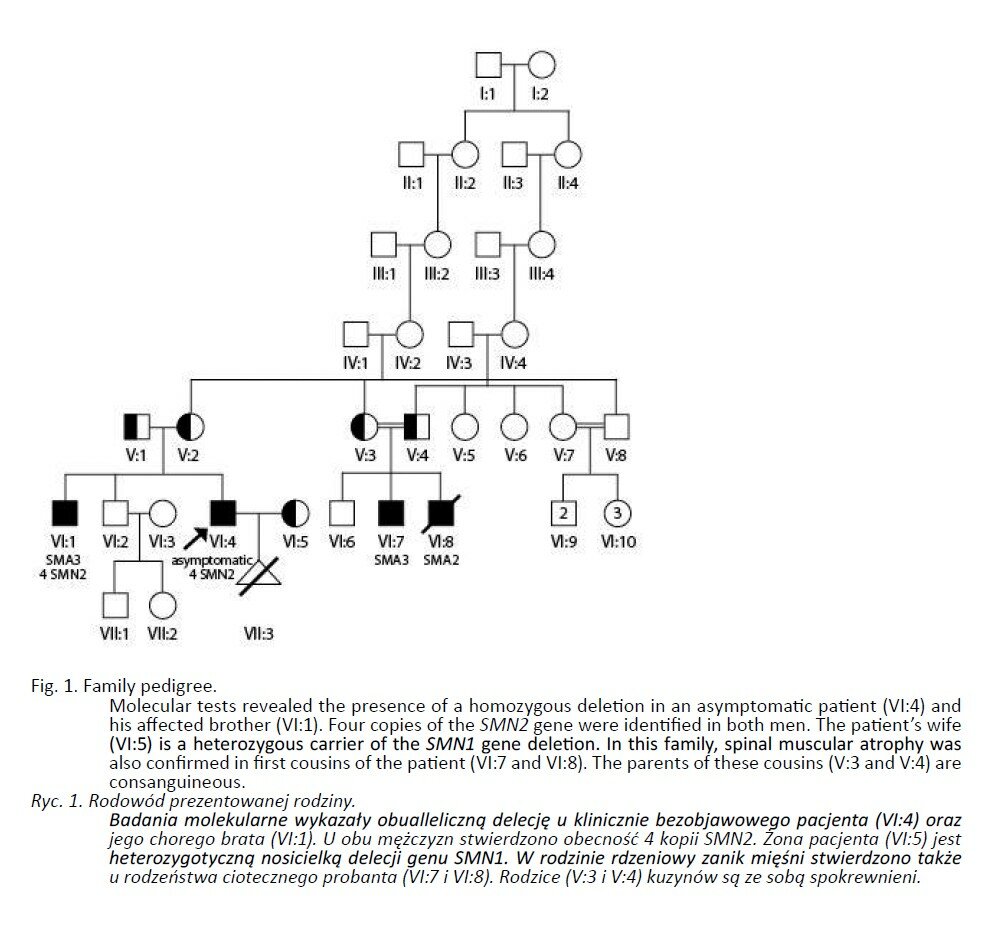
The couple were referred to a clinical geneticist seeking genetic advice about the risk of spinal muscular atrophy in their offspring. Both are healthy and non-consanguineous (the female aged 23 and 13 weeks pregnant, the male aged 27). Family history on the male side revealed SMA cases in relatives. The patient’s brother has a mild form of SMA (SMA3, Kugelberg-Welander). First symptoms appeared at age 7 and at age 26 the patient’s brother was unable to walk. He is now wheelchair-dependent. The same condition, albeit more severe, was diagnosed in two first cousins of the patient (Fig. 1). The younger of them had never attained the ability to walk and died at age 8 of respiratory insufficiency. The other cousin showed first symptoms at age 3 and later lost the ability to walk at age 15.
The clinical picture, results of electrophysiological studies and muscle biopsy all contributed to the affected individuals in the family being diagnosed with SMA, even though the condition has never been confirmed at the molecular level. The patient’s wife’s family history was irrelevant. The couple were given preliminary genetic advice. Assuming a carrier frequency in the general population of 1 in 35, the likelihood of SMA in the offspring was calculated as 1 in 210 (2/3x1/35x1/4)(10).
Blood was drawn from both the patient and his wife, in order to clarify their carrier status. The patient’s brother also underwent clinical assessment, and received molecular confirmation of the condition. For all molecular tests, DNA was isolated from blood samples by a simple salting-out procedure(24). All samples were collected on the basis of informed consent. The homozygous absence of exons 7 in the SMN1 gene was detected using restriction enzyme analysis, as described elsewhere (25). For SMN1 and SMN2 dosage analysis, the method described by Anhuf et al. was employed with the modification as described elsewhere (8, 26).
RESULTS
Molecular analyses revealed homozygous deletion in a clinically asymptomatic patient as well as in his affected brother. An asymptomatic patient did not show any symptoms on neurological examination. In both men, affected and asymptomatic- four copies of the SMN2 gene were detected. The patient’s wife was identified as a heterozygous carrier of the deletion, and further analyses revealed the presence of two copies of SMN2. The risk of SMA in the offspring of this couple was thus recalculated as 1 in 2 (50%). The patients were offered invasive prenatal testing (amniocentesis at 14 weeks, as described earlier) (27). The molecular analysis in the fetus revealed homozygous deletion of the SMN1 gene.
DISCUSSION
The identification of carrier status in the patient’s relatives provided a possibility of reliable genetic counselling. Parents of affected patients are obligatory carriers of the pathogenic SMN1 gene mutation. The de novo mutation rate in the SMN1 gene has been estimated as 2% (16). The risk of the condition in the offspring of mutation carriers is 25%. Thus, in high-risk SMA families there are indications as regards prenatal genetic testing in order to confirm or exclude the possibility of the disease in the fetus
With an increasing awareness of the SMA clinical picture and its consequences for families, other family members, such as partners, sibs, children or the more distant relatives of affected patients seek genetic consultation. Because of the autosomal recessive mode of inheritance, the risk of SMA in the above mentioned instances is usually low. Thus, for a carrier frequency of 1 in 35, it is about 1.4% (1x1/35x1/2) for the offspring of an affected person, 0.5% (2/3x1/35x1/4) for the offspring of a sib of an affected person and 0.4% (1/2x1/35x1/4) for a sib of parents of an affected person. As seen above, these risks overall are higher than population risk. In the family shown here the condition has been present not only in the patient’s brother, but also in his first cousins. Of interest is the fact that parents of first cousins are consanguineous, which increases the risk of an autosomal recessive condition in their offspring. The difficult issues of genetic counselling in SMA are further infl uenced by the incidence of asymptomatic individuals with a homozygous mutation in the SMN1 gene. The risk of SMA in the offspring of such persons, assuming the low risk of carrier status in their partners, is about 1.4%. In the presented family, the asymptomatic patient inherited the typical biallelic SMN1 gene mutation identical to the one detected in his brother. The patient’s wife has been identified as carrier of the deletion. Thus, the resulting risk of SMA in the offspring of this couple is 50% (1 in 2). Invasive prenatal testing of the fetus from her current pregnancy revealed the presence of a homozygous mutation. In the presented family, subsequent molecular analyses of the SMN1 gene verified the risk of SMA in the offspring.
Prior to the emergence of testing for carrier status, invasive prenatal diagnostics in SMA had mainly been offered to those couples at high risk of the disease developing in their offspring. The likelihood of SMA in other relatives of affected persons is 0.5-1%. Although this risk is considered low, it was until very recently sufficient to warrant invasive prenatal testing for couples at low risk as well. In Poland, five such tests had been performed up to 2004, each yielding normal results (27). Clearly, the introduction of carrier status testing limits indications for prenatal diagnostics to those couples at high risk only
Homozygous deletions in the SMN1 gene in clinically asymptomatic individuals are an extremely rare event. More than twenty such cases have been described in the literature to date (26). Based on several reports of quite large groups of relatives of affected persons, it is estimated that 0.5-0.7% of first-degree relatives have a typical nonpenetrant SMA mutation (26, 28, 29).This phenomenon still awaits plausible explanation. Asymptomatic parents with homozygous deletion usually have several (five to six) copies of the SMN2 gene, which compensates for the loss of SMN1. In healthy sibs of affected personsthis number is almost always the same, and the lack of symptoms in these individuals is most likely due to the interplay of modifying factors. Among the latter are the relatively well-known PLS3 gene, as well as epigenetic contributors (23, 30). Identification of PLS3 as a clinical modifier arises out of the observation that there is a marked predominance of female patients among asymptomatic homozygous deletion carriers. It is therefore conceivable that the infl uence of modifying factors is sex-linked. In 2008 Oprea et al. showed a greatly increased level of expression of PLS3 in asymptomatic women with homozygous SMN1 deletion, as compared with their affected sibs (30). The PLS3 gene located on an X chromosome plays an important role in neurogenesis, independent of SMN protein. The same authors have shown that changes in the pattern of methylation of SMN2 correlate positively with SMA symptoms (23). According to this study, patients with SMA3 have a significantly lower level of methylation (and higher SMN2 transcriptional activity) than SMA1 patients.
In the family presented, an asymptomatic patient has as many SMN2 copies as his affected brother. Although four copies are typically seen in mild forms of spinal muscular atrophy only, this cannot serve as an explanation for lack of symptoms. It seems that in this family another, so far unknown, modifying factor may exist. The presence of different forms of SMA (SMA 2 and 3) in first cousins (intrafamilial heterogeneity) may confirm the critical role of clinical modifiers.
In the patient’s wife only two copies of the SMN2 gene were detected, the number seen most commonly in the general population, as well as in 40-45% of controls (7, 20). These estimates have been reproduced in the Polish population, in which 43% (262 of 600) of those examined carried two copies and 41% (250 of 600) only a single copy of SMN2 gene (10).
We could not make an estimate of the number of SMN2 copies in the fetus in the family shown here. However, it can be assumed that it inherited one copy of the SMN2 gene from the mother and two copies from the father. Quantitative analyses could not assess the allelic distribution of these copies either. Potentially, the fetus could have inherited three copies, consistent with the forms of the disease that are severe (41% of SMA1 patients) or intermediate (84% of SMA2 patients) (22). However, the number of SMN2 copies must not be recognized as a prognostic factor, since three SMN2 copies are to be identified in both mildly and severely affected patients.
CONCLUSIONS
The above observations point to the necessity of competent and reliable genetic counselling to be provided in SMA-risk families. They also indicate the need of secondary preventative measures to be introduced. This becomes even more important in the context of the relatively high frequency of carriers of the SMN1 gene deletion in the general population (1 in 35). Another clinically significant, although rare, factor is asymptomatic homozygous deletion of the SMN1 gene. The introduction of diagnostic tests allowing for the confirmation of SMA or the identification of mutation carriers will eventually lead to both improved and more individualized medical care, as well as the provision of reliable genetic counselling.
REFERENCES
1. Spiegler A.W., Hausmanowa-Petrusewicz I., Borkowska J.: Population data on acute infantile and chronic childhood spinal muscular atrophy in Warsaw. Hum. Genet., 1990, 85, 211-214.
2. Thieme A., Mitulla B., Schulze F., Spiegler A.W.: Epidemiological data on Werdnig-Hoffmann disease in Germany (West-Thuringen). Hum. Genet. 1993, 91, 295- 297.
3. Thieme A., Mitulla B., Schulze F., Spiegler A.W.: Chronic childhood spinal muscular atrophy in Germany (West- Thüringen) – an epidemiological study. Hum. Genet., 1994, 93, 344-346.
4. Kvasnicova M., Stykova J., Hudec P.: Incidence of spinal muscular atrophy and Duchenne muscular atrophy in child population of middle Slovakia. Bratisl. Lek. Listy, 1994, 95, 78-82.
5. Smith M., Calabro V., Chong B., Gardiner N., Cowie S., du Sart D.: Population screening and cascade testing for carriers of SMA. Europ. J. Hum. Genet., 2007, 15, 759-766.
6. Cusin V., Clermont O., Gerard S., Chantereau D., Elion J.: Prevalence of SMN1 deletion and duplication in carrier and normal populations: implication for genetic counselling. J. Med. Genet., 2003, Epub 40(4), e39.
7. Feldkötter M., Schwarzer V., Wirth R., Wienker T.F., Wirth B.: Quantitative analysis of SMN1 and SMN2 based on real-time Light Cycler PCR: Fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet., 2002, 70, 358-368.
8. Anhuf D., Eggermann T., Rudnik-Schöneborn S., Zerres K.: Determination of SMN1 and SMN2 copy number using TaqMan Technology. Hum. Mut., 2003, 22, 74-78.
9. Hendrickson B.C., Donohoe C., Akmaev V.R., Sugarman E.A., Labrousse P., Boguslavskiy L. Flynn K., Rohlfs E.M., Walker A., Allitto B., Sears C., Scholl T.: Differences in SMN1 allele frequencies among ethnic groups within North America. J. Med. Genet, 2009, 46, 641-644.
10. Jędrzejowska M., Milewski M., Zimowski J., Zagożdżon P., Kostera-Pruszczyk A., Borkowska J., Sielska D., Jurek M., Hausmanowa-Petrusewicz I.: SMA incidence in Poland – more frequent than predicted? Neuroepidemiology 2010, 34, 152-157.
11. Munsat T., Davies K.: Report of International SMA Consortium Meeting. Neuromusc. Disord., 1992, 2, 423- 428.
12. Brahe C., Servidei S., Zappata S., Ricci E., Tonali P., Neri G.: Genetic homogeneity between childhood-onset and adult-onset autosomal recessive spinal muscular atrophy. Lancet, 1995, 346, 741-742.
13. Macleod M.J., Taylor J.E., Lunt P.W., Mathew Ch.G., Robb S.A.: Prenatal onset spinal muscular atrophy. Europ. J. Paediatr. Neurol., 1999, 3, 65-72.
14. Hahnen E., Forkert R., Marke Ch., Rudnik-Schöneborn S., Schönling J., Zerres K., Wirth B.: Molecular analysis of candidate genes on chromosome 5q13 in autosomal recessive spinal muscular atrophy: evidence of homozygous deletions of the SMN gene in unaffected individuals. Hum. Mol. Genet., 1995, 4, 1927-1933.
15. Cobben J.M., Van der Steege G., Grootscholten P., De Visser M., Scheffer H., Buys C.: Deletions of the survival motor neuron gene in unaffected siblings of patients with spinal muscular atrophy. Am. J. Hum. Genet., 1995, 57, 805-808.
16. Lefebvre S., Burglen L., Roboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M. et al.: Identification and characterization of spinal muscular atrophy determining gene. Cell 1995, 80, 155- 165.
17. Jędrzejowska M., Wiszniewski W., Zimowski J., Kostera- Pruszczyk A., Ryniewicz B., Bal J., Zaremba J., Mazurczak T., Hausmanowa-Petrusewicz I.: Application of a rapid non-invasive technique in the molecular diagnosis of spinal muscular atrophy (SMA). Neur. Neurochir. Pol., 2005, 39, 89-94.
18. Wirth B.: An update mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy(SMA). Hum. Mutat., 2000, 15, 228-237.
19. Monani U.R., Sendtner M., Coovert D.D., Parsons D.W., Andreassi C., Le T.T., Jablonka S., Schrank B., Rossol W., Prior T.W., Morris G.E., Burghes A.H.: The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(-/-) mice and results in a mouse with spinal muscular atrophy. Hum. Mol. Genet., 2000, 9, 333- 339.
20. Mailman M.D., Heinz J.W., Audrey C.P., Snyder P.J., Sedra M.S., Wirth B., Burghes A.H.M., Prior T.W.: Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet. Med., 2002, 4, 20-26.
21. Wirth B., Brichta L., Schrank B., Lochmuuler H., Blick S., Baasner A., Heller C.: Mildly affected patients with spinal muscular atrophy are partially protected by increased SMN2 copy number. Hum. Genet., 2006, 119, 422-428.
22. Jędrzejowska M., Milewski M., Zimowski J., Borkowska J., Kostera-Pruszczyk A., Sielska D., Jurek M., Hausmanowa- Petrusewicz I.: Phenotype modifiers of spinal muscular atrophy: the number of SMN2 gene copies, deletion in the NAIP gene and probably gender infl uence the course of the disease. Acta Biochim. Pol., 2009, 56, 103-831.
23. Hauke J., Riessland M., Lunke S., Eyüpoglu I.Y., Blümcke I., El-Osta A., Wirth B., Hahnen E.: Survival motor neuron gene 2 silencing by DNA methylation correlates with spinal muscular atrophy disease severity and can be bypassed byhistone deacetylase inhibition. Hum. Mol. Genet., 2009, 18, 304-317.
24. Miller S.A., Dykes D.D., Poleski H.F.: A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic. Acid Res., 1988, 16, 1215.
25. Scheffer H., Cobben J.M., Matthijs G., Wirth B.: Best practice guidelines for molecular analysis in spinal muscular atrophy. Europ. J. Hum. Genet., 2001, 9, 484-491.
26. Jędrzejowska M., Borkowska J., Zimowski J., Kostera- Pruszczyk A., Milewski M., Jurek M., Sielska D., Kostyk E., Nyka W., Zaremba J., Hausmanowa-Petrusewicz I.: Unaffected patients with a homozygous absence of the SMN1 gene. Europ. J. Hum. Genet., 2008, 16, 930-934.
27. Jędrzejowska M., Zimowski J., Wiszniewski W., Sielska D., Bal J., Mazurczak T., Hausmanowa-Petrusewicz I., Zaremba J.: Prenatal diagnosis of spinal muscular atrophy (SMA) – indications, restrictions, interpretation of results. Med. Wieku Rozwoj., 2004, 8, 651-661.
28. Prior T.W., Swoboda K.J., Scott H.D., Hejmanowski A.Q.: Homozygous SMN1 deletions in unaffected family members and modification of the phenotype by SMN2. Am. J. Med. Genet., 2004, A130A, 307-310.
29. Wang C.H., Xu J., Carter T.A., Ross B.M., Dominski M.K., Bellcross C.A., Penchaszadeh G.K., Munsat T.L., Gilliam T.C.: Characterization of survival motor neuron (SMNT) gene deletions in asymptomatic carriers of spinal muscular atrophy. Hum. Mol. Genet., 1996, 5, 359-365.
30. Oprea G.E., Kröber S., McWhorter M.L., Rossoll W., Müller S., Krawczak M., Bassell G.J., Beattie C.E., Wirth B.: Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science, 2008, 320, 524-527.
Adres do korespondencji:
Maria Jędrzejowska
Zespół Nerwowo-Mięśniowy
Instytut Medycyny Doświadczalnej i Klinicznej
im. M. Mossakowskiego
Polska Akademia Nauk
ul. Pawińskiego 5, 02-106 Warszawa
tel./fax (22) 60-86-631
[email protected]